Blood vessel cells implicated in chronic inflammation of obesity
DALLAS – Dec. 29, 2020 – When fat cells in the body are stuffed with excess fat, the surrounding tissue becomes inflamed. That chronic, low-level inflammation is one of the driving factors behind many of the diseases associated with obesity. Now, UT Southwestern scientists have discovered a type of cell responsible, at least in mice, for triggering this inflammation in fat tissue. Their findings, published in Nature Metabolism, could eventually lead to new ways to treat obesity.

“The inflammation of fat cells in obese individuals is linked to many of the comorbidities we associate with being overweight – cancer, diabetes, heart disease, and infection,” says study leader Rana Gupta, Ph.D., associate professor of internal medicine. “By identifying these cells, we’ve taken a step toward understanding some of the initial events that contribute to that inflammation.”
When a person consumes more calories than needed, the excess calories are stored in the form of triglycerides inside fat tissue, also known as white adipose tissue (WAT). Researchers know that in obese people, WAT becomes overworked, fat cells begin to die, and immune cells become activated. But the exact mechanism by which this inflammation occurs isn’t fully understood.
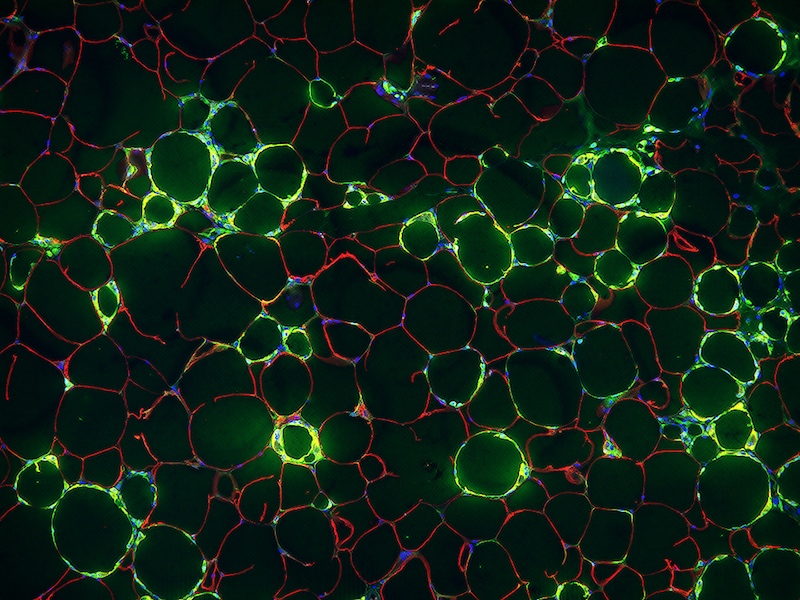
While many studies have focused on the signaling molecules produced by the fat cells or immune cells in WAT that might contribute to inflammation, Gupta’s team took a different approach. They focused instead on the vessels that carry blood – as well as immune cells and inflammatory molecules – into WAT.
In 2018, Gupta and his colleagues identified a new type of cell lining these blood vessels in mice – an adipose progenitor cell (APC), or precursor cell that goes on to generate mature fat cells. But unlike most APCs, the new cells – dubbed fibro-inflammatory progenitors, or FIPs – produced signals that encouraged inflammation. In the new work, the researchers looked more closely at the role of the FIPs in mediating inflammation.
Within just one day of switching young male mice to a high-fat diet, Gupta and his colleagues discovered that the FIPs quickly increased the number of inflammatory molecules produced. After 28 days on a high-fat diet, they found a substantial increase in the proportion of FIPs compared with other APCs.
“This is the first study to demonstrate that these cells play a very active, early role in being gatekeepers of inflammation in fat tissue,” says Gupta.
To show that the increase in the number and activity of the FIPs was not just a side effect of already-inflamed fat cells, the team removed a key immune signaling gene, Tlr4, from the FIPs in some mice. After five months on a high-fat diet, the mice lacking Tlr4 had gained just as much weight, and just as much fat, as other mice on a high-fat diet. But the genetically engineered mice – with FIPs that could no longer generate the same signals – no longer had high levels of inflammation. Instead, the levels of inflammatory molecules in their WAT were closer to the levels seen in mice on low-fat diets.
Gupta and his colleagues went on to show that increasing levels of a related signaling molecule, ZFP423, in FIPs can also ameliorate the inflammation in mouse fat cells. The findings point toward possible avenues to pursue to lower the risk of disease in people with obesity.
“It looks like ZFP423 could be an important brake in terms of slowing the inflammatory signals in these cells,” says Gupta. “Of course, it remains to be seen if that’s true in humans as well as mice.”
Gupta’s group is planning future experiments to better understand what aspect of a high-fat diet initiates the increased inflammatory signaling in FIPs, as well as whether the results hold true in human fat.
Other UTSW researchers who contributed to this study were Bo Shan, Mengle Shao, Qianbin Zhang, Chelsea Hepler, Vivian Paschoal, Spencer Barnes, Lavanya Vishvanath, Yu An, Lin Jia, Venkat Malladi, Douglas Strand, Olga Gupta, Joel Elmquist and Dayoung Oh.
This research was supported by funds from the National Institutes of Health (NIDDK F31DK113696, R01 DK104789, R56 DK119163, R01 DK119163, R01 DK115477, R01 DK108773, R03 DK101865, PO1 DK088761 and NIAAA K01AA024809), the American Heart Association (16POST26420136 and 14SDG19880020), the American Diabetes Association (1-18-PMF-030) and the Cancer Prevention and Research Institute of Texas (RP150596).